
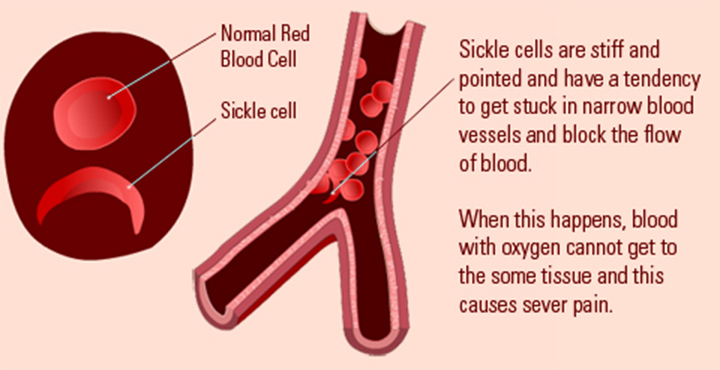
What is sickle cell anaemia?
Sickle cell anaemia (sickle cell disease) is a disorder of the blood caused by inherited abnormal haemoglobin (the oxygen- carrying protein within the red blood cells). The abnormal haemoglobin causes distorted (sickled) red blood cells.
The sickled red blood cells are fragile and prone to rupture. When the number of red blood cells decreases from rupture (haemolysis), anaemia is the result. This condition is referred to as sickle cell anaemia. The irregular sickled cells can also block blood vessels causing tissue and organ damage and pain. Sickle cell anaemia is one of the most common inherited blood anaemia’s. The disease primarily affects Africans and African Americans. It is estimated that in the United States, some 90,000 to 100,000 Americans are afflicted with sickle cell anaemia. Overall, current estimates are that one in 500 African American births is affected with sickle cell anaemia.
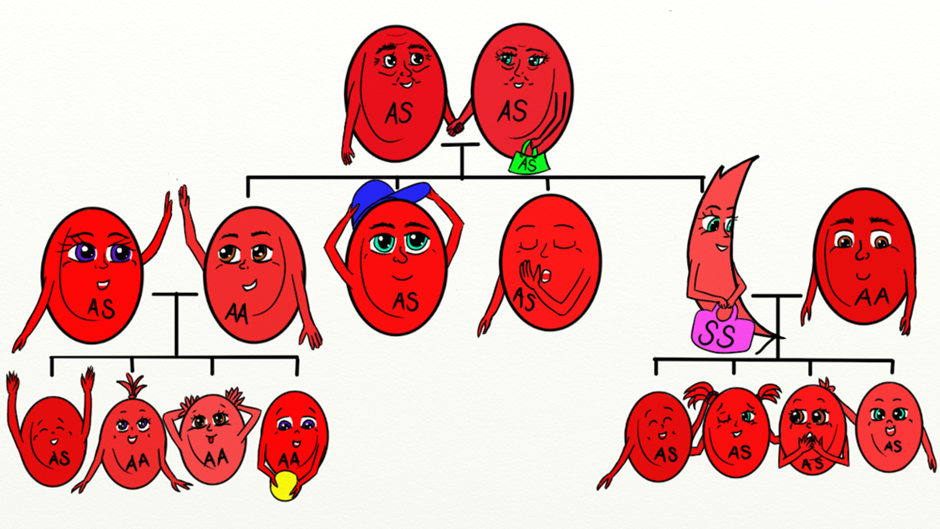
How is sickle cell anaemia inherited?
Sickle cell anaemia is inherited as an autosomal (meaning that the gene is not linked to a sex chromosome) recessive condition. This means that the gene can be passed on from a parent carrying it to male and female children. In order for sickle cell anaemia to occur, a sickle cell gene must be inherited from both the mother and the father, so that the child has two sickle cell genes. The inheritance of just one sickle gene is called sickle cell trait or the “carrier” state. Sickle cell trait does not cause sickle cell anaemia. Persons with sickle cell trait usually do not have many symptoms of disease and have hospitalization rates and life expectancies identical to unaffected people. When two carriers of sickle cell trait mate, their offspring have a one in four chance of having sickle cell anaemia. (In some parts of Africa, one in five persons is a carrier for sickle cell trait.
 Haemoglobin is an iron containing globular protein in red blood cells that transports oxygen. It consists of two alpha or alpha-like polypeptide chains, each of which pairs with a beta, gamma or delta chain. Over 1,600 variants of haemoglobin molecules have been reported and named based on their mutations as well as geographical locations of where they are prevalent. The haemoglobin molecule present predominantly in mature circulation is Haemoglobin A (HbA). A single nucleotide mutation (GAG to GTG) causes the substitution of the amino acid valine for glutamic acid at the 6th position of the beta chain in Haemoglobin A, resulting in Haemoglobin S (HbS). A GAG to AAG mutation causes the substitution of lysine for glutamic acid at the same position, resulting in Haemoglobin C (HbC). Absence or decreased synthesis of the beta chain results in beta-thalassemia and a heterozygous genotype S- Thal. Although relatively rare in North America, this mutation also causes sickle beta-thalassemia. It has been well documented that the SS genotype causes more severe systemic disease than SC and S-Thal genotypes, while the SC and S-Thal genotypes present with a greater incidence of ocular manifestations, including proliferative sickle cell retinopathy. The underlying systemic and ocular manifestations of SCD (Sickle Cell Disease) are a result of vaso-occlusive ischemia due to the blocking of blood vessels by sickle-shaped erythrocytes.
Haemoglobin is an iron containing globular protein in red blood cells that transports oxygen. It consists of two alpha or alpha-like polypeptide chains, each of which pairs with a beta, gamma or delta chain. Over 1,600 variants of haemoglobin molecules have been reported and named based on their mutations as well as geographical locations of where they are prevalent. The haemoglobin molecule present predominantly in mature circulation is Haemoglobin A (HbA). A single nucleotide mutation (GAG to GTG) causes the substitution of the amino acid valine for glutamic acid at the 6th position of the beta chain in Haemoglobin A, resulting in Haemoglobin S (HbS). A GAG to AAG mutation causes the substitution of lysine for glutamic acid at the same position, resulting in Haemoglobin C (HbC). Absence or decreased synthesis of the beta chain results in beta-thalassemia and a heterozygous genotype S- Thal. Although relatively rare in North America, this mutation also causes sickle beta-thalassemia. It has been well documented that the SS genotype causes more severe systemic disease than SC and S-Thal genotypes, while the SC and S-Thal genotypes present with a greater incidence of ocular manifestations, including proliferative sickle cell retinopathy. The underlying systemic and ocular manifestations of SCD (Sickle Cell Disease) are a result of vaso-occlusive ischemia due to the blocking of blood vessels by sickle-shaped erythrocytes.
Epidemiology
In African-Americans in North America, the incidence of sickle cell trait (AS) is about 8%, while that of SCD is 0.4%. The incidence of SC, AC and S-Thal genotypes among African-Americans in North America is 0.2%, 2% and 0.03%, respectively. Importantly, the incidence of proliferative retinopathy is highest in patients with SC or S-Thal (33% and 14% respectively), while patients with SS have a 3% incidence of proliferative retinopathy.
Pathophysiology
Normal blood cells that are round and oval can easily pass through smaller blood vessels including capillaries. However, local hypoxic conditions alter the shape of the red cells in SCD patients, leading to rigid, sickle-shaped red blood cells (RBCs), due to the irreversible conversion of soluble haemoglobin into crystalline haemoglobin. Changes in the adhesive properties of capillary endothelial cells due to hypoxia lead to deceased vascular flow and occlusion of vessels. This trapping of sickle-shaped red cells in the small blood vessels in various structures of eye, both in the anterior and posterior segments, leads to characteristic damage. The clinical manifestations vary depending on the presence or absence of vaso-proliferative changes. Hence, this disease can be classified as;
- Non-proliferative sickle cell retinopathy (NPSR) and
- Proliferative sickle cell retinopathy (PSR).
Non-proliferative Sickle retinopathy (NPSR):
The retinal changes in NPSR occurs secondary to vaso-occlusion and local ischemia. Occlusion of retinal vasculature first appears in the peripheral retina as “salmon patches”, which represent retinal haemorrhage from superficial blood vessels. These lesions are round or oval in shape and can be elevated or flattened. Although they are initially red, they become salmon-coloured due to the haemolysis of red blood cells. Over time, the haemorrhage is resorbed and the area appears normal with refractile deposits, which are iridescent spots of hemosiderin and macrophage deposition just beneath the internal limiting membrane. Migration and proliferation of retinal pigment epithelium (RPE) leads to the development of black sunburst spots which likely occur in response to haemorrhage. Reduction in visual acuity is usually from the occlusion of perifoveal capillaries, causing a concave macular depression from RPE degeneration. Occlusions in the choroidal circulation with associated breaks in Bruch’s membrane are known to cause angioid streaks, a phenomenon seen in many retinal diseases including NPSR and PSR.
Proliferative Sickle Retinopathy (PSR):
As noted above, the ocular complications of PSR are more characteristically seen in HbC (SC) and SThal disease, rather than in SS disease. As localized vascular occlusion leads to the changes described above in NSPR, chronic changes of local hypoxia and ischemia lead to the upregulation of vascular growth factors, which may result in retinal neovascularization, pre-retinal or vitreous haemorrhage and tractional retinal detachment.
Goldberg classified PSR into 5 different stages;
- Stage I Peripheral arterial occlusion.
- Stage II Peripheral arteriovenous anastomoses, representing dilated pre-existing capillaries (hairpin loop).
- Stage III Neovascular and fibrous proliferation (sea fan) occurring at the posterior border of non-perfusion. A subsequent White Sea fan appearance is due to auto-infarction of the neovasculature.
- Stage IV Vitreous haemorrhage.
- Stage V Tractional retinal detachment.
While extra-retinal fibrovascular proliferation is seen in other disease entities, most commonly diabetic retinopathy, neovascularization in PSR typically begins peripherally, rather than centrally as in diabetic retinopathy. Visual loss in PSR occurs most commonly due to vitreous haemorrhage and tractional retinal detachment.
Clinical Signs/Symptoms
Vaso-occlusion of conjunctival vessels leads to the development of “comma” shaped vessels due to the accumulation of sickled RBCs at the distal end of the capillaries.
Vascular changes in the optic disc are transient, but present as dark red lesions also visible as dilated dark capillary vessels on fluorescein angiography.
Patients may see flashes, floaters or dark shadows, which may be indicative of vitreoretinal traction or detachment.
Diagnosis
- History of sickle cell disease.
- Presence of comma-shaped blood vessels in the bulbar conjunctiva (“comma sign”) and angioid streaks on funduscopic examination.
- Fluorescein angiography to examine blood flow in retina and choroid.
- Detailed structural examination with spectral-domain optical coherence tomography (SD-OCT).
Differential Diagnosis
Sickle cell retinopathy should be distinguished from the conditions listed below. A prior history of sickle cell disease helps rule out many of these conditions, although it is possible for these conditions to occur simultaneously with NPSR/PSR.
- Diabetic retinopathy.
- Retinal vascular occlusion.
- Talc emboli.
- Hypertensive retinopathy.
- Sarcoidosis.
- Eale’s disease.
- Infectious diseases (e.g. dengue).
- Ocular ischemic syndrome.
- Retinopathy of prematurity.
- Familial exudative vitreoretinopathy.
- Chronic myelogenous leukaemia.
- Scleral buckle.
- Hyperviscosity syndrome.
Management
The goal of therapy for NPSR/PSR is to prevent development of ischemia, infarction and the dreaded complication of neovascularization. Complete ophthalmic examination twice a year is recommended for sickle cell patients (SC, SS, S-thal, etc).
Baseline fluorescein angiography may be performed to examine blood flow in the retina and choroid. Since humans have an inherent ability to produce foetal haemoglobin, emerging therapies that stimulate foetal haemoglobin have been shown to be beneficial. For example, hydroxycarbamide treatment has been demonstrated to have a beneficial effect in children in preventing sickle cell retinopathy.
Similarly, reducing HbSS red blood cells by exchange transfusion has also shown to be beneficial. Hyperbaric oxygen therapy may reverse the pathology and improve visual acuity.
As complications of PSR arise, intravitreal administration of anti-vascular endothelial growth factor (anti-VEGF) therapy and/or scatter laser photocoagulation may lead to regression of neovascularization. The high incidence of neovascularization and spontaneous regression complicates treatment modalities. Feeder vessel photocoagulation to close sea fan structures with argon laser over Xenon arc is preferred as Xenon arc tends to damage Bruch’s membrane, thus increasing the risk of choroidal neovascularization.
Similarly, 360 degrees scatter photocoagulation of the peripheral retina has been shown to reduce the number of sea fan structures due to neovascularization, although a protective effect is better with feeder vessel photocoagulation. In the event of vitreous haemorrhage, patients need to be examined at regular intervals to document clearance and resolution of visual acuity. In cases of non-clearing vitreous haemorrhage, pars plana vitrectomy remains the treatment of choice, despite the higher risk of intraoperative and postoperative complications associated with PPV in patients with sickle cell retinopathy compared to patients without SC or S-Thal.
Complications and contraindications
Due to the risk of retinal detachment post-treatment, scatter photocoagulation is the preferred method of intervention to cause regression of sea fan structures if the sea fans do not auto-infarct. However, retinal tear and rhegmatogenous retinal detachments may occur after scatter photocoagulation.
The rate of surgical complications in patients with PSR has been reported to be as high as 50% for traction/rhegmatogenous retinal detachment in a recent study.
Presence of traumatic hyphema in African Americans warrants testing the patients for sickle cell haemoglobinopathies due to ocular complications that can result from sickle red blood cells.
To control intraocular pressure, use of carbonic anhydrase inhibitors, especially acetazolamide, is contraindicated as it induces systemic acidosis and increases the local concentration of ascorbic acid in the aqueous humour; hence, the resulting low pH enhances sickling of RBCs. Instead of acetazolamide, the use of methazolamide is recommended. Scleral buckling surgery also carries higher intra- and post-operative risk with similar complications as pars plana vitrectomy in patients with sickle cell disease.